
Friday, December 7, 2018
NMR and the Taste of Christmas - Gingerbread
The holiday season is full of delicious treats. Aside from rum spiked eggnog, candy canes, fruitcake, shortbread and cranberry sauce, one of my favorite Christmas treats is gingerbread. Whether you enjoy biting the limbs off a gingerbread man or munching on the roof of a gingerbread house, you cannot escape the wonderful aroma and flavor of ginger and cinnamon. These fragrant spices can be easily examined by NMR spectroscopy. The figure below shows the 600 MHz 1H NMR spectra of CDCl3 extracts of ground ginger (top) and ground cinnamon (bottom).
 The main constituents of these extracts are 6-gingerol and cinnamaldehyde for the ginger and cinnamon extracts, respectively. Think of these compounds and their NMR spectra while you bite the head off your next gingerbread man in front of your beautifully decorated Christmas tree. Merry Christmas!
The main constituents of these extracts are 6-gingerol and cinnamaldehyde for the ginger and cinnamon extracts, respectively. Think of these compounds and their NMR spectra while you bite the head off your next gingerbread man in front of your beautifully decorated Christmas tree. Merry Christmas!
Tuesday, November 20, 2018
NOAH - Faster 2D Data Collection
NMR users typically run 1H, 13C, COSY, HSQC, HMBC and NOESY spectra to elucidate the structures of small molecules. Even with cryogenically cooled probes and pulsed field gradient accelerated methods, collecting 2D spectra can be quite time consuming. For concentrated samples, each 2D experiment will typically take minutes to tens of minutes to collect. Much of this time is the result of waiting for T1 relaxation in each of the experiments. Recently, Kupce and Claridge1,2 have developed a technique using standard NMR hardware where multiple 2D methods are concatenated in a single super pulse sequence employing a single relaxation delay. They have called the technique NOAH (NMR by Ordered Acquisition using 1H detection) The time saving of the NOAH technique compared to individually collected 2D spectra results from waiting a single relaxation delay for all experiments rather than a single relaxation delay for each separately acquired spectrum. The data for each spectrum is acquired in separate memory blocks which are separated after data collection allowing the data for each 2D method to be processed individually. The data can also be processed in automation. The authors have kindly made this method accessible to all Bruker users through the Bruker User Library which contains pulse sequences, parameter sets, automation scripts and detailed instructions. The left-hand panel of the figure below shows the 600 MHz HMBC, Ed-HSQC and COSY spectra obtained from the NOAH-3 BSC (HMBC, HSQC, COSY) pulse sequence for sucrose in DMSO-d6. The right-hand panel shows separately acquired 2D data sets for comparison.
 The NOAH spectra were obtained from the raw concatenated data with the automation script provided. The high quality NOAH-3 data using 2 scans, 256 increments and a 2 second relaxation delay, took only 24 minutes to acquire in comparison to the separately acquired 2D spectra obtained with similar parameters, which took a total of 59 minutes to acquire. This represents a time saving of 35 minutes or 59%. It should also be noted that the data from the NOAH-3 BSC sequence is of comparable quality to that of the individually collected spectra.
The NOAH spectra were obtained from the raw concatenated data with the automation script provided. The high quality NOAH-3 data using 2 scans, 256 increments and a 2 second relaxation delay, took only 24 minutes to acquire in comparison to the separately acquired 2D spectra obtained with similar parameters, which took a total of 59 minutes to acquire. This represents a time saving of 35 minutes or 59%. It should also be noted that the data from the NOAH-3 BSC sequence is of comparable quality to that of the individually collected spectra.
1. Eriks Kupce and Tim D. W. Claridge. Chem. Commun. 54, 7139 (2018).
2. Eriks Kupce and Tim D. W. Claridge. Angew. Chem. Int. Ed., 56, 11779 (2017).

1. Eriks Kupce and Tim D. W. Claridge. Chem. Commun. 54, 7139 (2018).
2. Eriks Kupce and Tim D. W. Claridge. Angew. Chem. Int. Ed., 56, 11779 (2017).
Wednesday, October 31, 2018
Pure-Shift HSQC
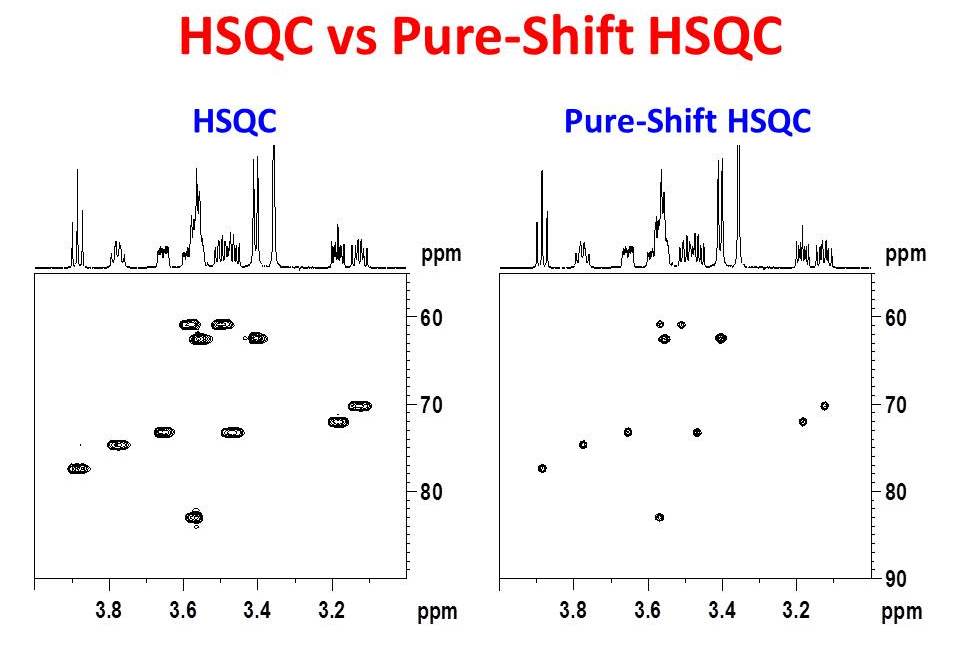
Pure-shift NMR has become more and more common over the last few years. A special issue of Magnetic Resonance in Chemistry has recently been dedicated to developments in these methods. Pure-shift NMR methods offer simplified proton NMR spectra free of 1H - 1H coupling. These methods have been extended to proton detected 2D NMR measurements, yielding 2D data sets with higher proton resolution compared to conventional 2D measurements. The NMR Methodology Group at the University of Manchester has been a primary contributor to this technique and has kindly shared their efforts on-line. The figure below compares the 600 MHz partial Pure-Shift HSQC spectrum of sucrose in DMSO-d6 to a more conventional HSQC spectrum acquired under similar conditions. The projections on the spectra are independently collected high resolution 1H NMR spectra. Clearly, the Pure-Shift HSQC data have higher 1H resolution than the more conventional HSQC. What may not be so obvious from the figure is that the sensitivity is also improved in the Pure-Shift HSQC. The gain in signal-to-noise-ratio depends strongly on the degree of coupling collapsed. For some signals in this spectrum, an improvement in the signal-to-noise-ratio as high as 72% was observed.
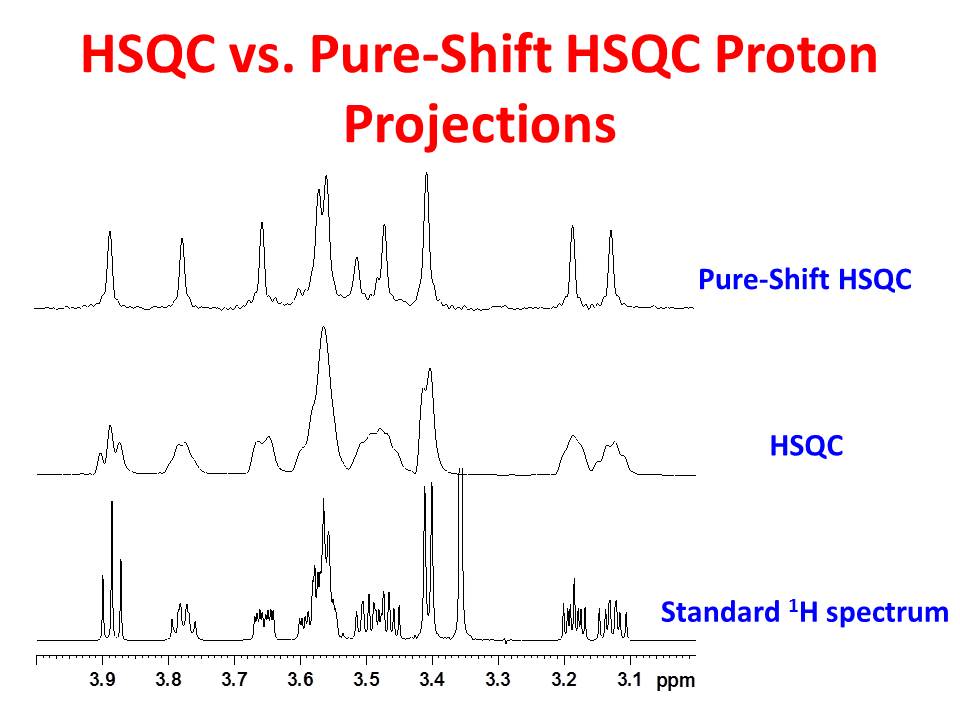
 The the top and middle panels of the figure below show the 1D 1H projections of the Pure-Shift HSQC and HSQC data from the above figure, respectively. The bottom panel is the conventional high resolution 1H spectrum for comparison.
The the top and middle panels of the figure below show the 1D 1H projections of the Pure-Shift HSQC and HSQC data from the above figure, respectively. The bottom panel is the conventional high resolution 1H spectrum for comparison.
 Clearly, the Pure-Shift HSQC proton projection offers much improved resolution.
Clearly, the Pure-Shift HSQC proton projection offers much improved resolution.
Thursday, August 23, 2018
13C Satellite Observation with 1D 1H - 13C HSQC
Two dimensional 1H - 13C or 1H - 15N HSQC spectra are typically used to obtain one-bond heteronuclear correlation information between 1H and 13C or 1H and 15N. The data are proton detected and typically employ pulsed field gradients for coherence selection. The technique uses an incremented delay for chemical shift correlation and essentially discards the very intense signals from protons bound to 12C or 14N while enhancing the remaining doublet signals for protons bound to 13C or 15N. Usually the 1H FIDs are collected with 13C or 15N decoupling to collapse the doublets into singlets in the F2 domain leaving a single correlation between proton signals in F2 at the 13C or 15N chemical shifts in F1 of the nuclei to which the protons are bound. One can use a 1D version of this experiment (without the incremented delay for chemical shift correlation) to collect 1H NMR spectra exclusively of the protons bound to 13C or 15N. If used without 13C or 15N decoupling, it allows easy observation of the 13C or 15N satellites in the absence of the very intense signals due to protons bound to 12C or 14N and allows the precise measurement of one-bond 1H - 13C or 1H - 15N coupling constants. The top panel of the figure below shows the 300 MHz 1D 1H - 13C HSQC spectrum of ethyl acetate. Clearly the 13C satellites are observed and the 1H - 12C signals are suppressed. The 1H spectrum of ethyl acetate is shown in the bottom panel of the figure for comparison.


Thursday, June 21, 2018
Glycine as a 13C CPMAS Setup Sample
Glycine is an excellent setup compound for 13C CPMAS NMR measurements. Its utility in this regard has been described in detail.1,2 It can easily be observed in one scan and has reasonably short 1H T1's, allowing it to be used for 1H 90° pulse calibration and to setup the Hartmann Hahn matching condition. The width of the methylene carbon signal can be conveniently used to evaluate the proton decoupling efficiency. The width and shape of the carbonyl signal are very sensitive to the angle at which the sample is spun and can be used to set the magic angle with a reasonably high degree of precision. In addition the carbonyl resonance is sharp and can be used as a secondary standard for chemical shift calibration. If used as a secondary chemical shift standard, one must be aware that glycine has three polymorphic forms, each with different carbonyl chemical shifts. The polymorphic form is not generally displayed on the reagent bottle and different suppliers may provide different polymorphs or mixtures of polymorphs. It is therefore important to know which polymorph is being used to calibrate the chemical shift scale. The α and γ polymorphs are the most common and stable, while the β polymorph is less stable and easily converted over time to the α polymorph. Furthermore, the β polymorph has a very short 1H T1ρ at room temperature and therefore difficult to observe with typical millisecond CP contact times. The γ polymorph can be converted to the α polymorph at 165°C. The chemical shifts for the carbonyl resonances for the α and γ polymorphs are 176.5 ppm and 174.6 ppm, respectively.1 The chemical shift of the β polymorph is between that of the α and γ polymorphs however, it is not usually observed. The figure below shows the carbonyl region of the 13C CPMAS spectrum of three samples of glycine: pure α, pure γ and a mixture of the α and γ polymorphs.
 If a single carbonyl resonance is observed for a sample of glycine using typical millisecond CP contact times, one can determine if it is the α or γ polymorph by measuring its chemical shift with respect to another chemical shift standard. Alternatively, since the 1H T1ρ characteristics for the α and γ polymorphs are quite different from one another at room temperature, the authors of reference 1 report that a CPMAS spectrum collected with a 20 msec contact time will show almost no signal for the carbonyl carbon of the γ polymorph. The carbonyl signal of the α polymorph, on the other hand, will be only slightly attenuated compared to a CPMAS spectrum measured with a 1 msec contact time.
If a single carbonyl resonance is observed for a sample of glycine using typical millisecond CP contact times, one can determine if it is the α or γ polymorph by measuring its chemical shift with respect to another chemical shift standard. Alternatively, since the 1H T1ρ characteristics for the α and γ polymorphs are quite different from one another at room temperature, the authors of reference 1 report that a CPMAS spectrum collected with a 20 msec contact time will show almost no signal for the carbonyl carbon of the γ polymorph. The carbonyl signal of the α polymorph, on the other hand, will be only slightly attenuated compared to a CPMAS spectrum measured with a 1 msec contact time.
1. M.J. Potrzebowski, P. Tekely, Y. Dusausoy. Solid State Nuclear Magnetic Resonance. 11, 253 (1998).
2. R. E. Taylor. Concepts in Magnetic Resonance. 22A, 1 (2004).

1. M.J. Potrzebowski, P. Tekely, Y. Dusausoy. Solid State Nuclear Magnetic Resonance. 11, 253 (1998).
2. R. E. Taylor. Concepts in Magnetic Resonance. 22A, 1 (2004).
Wednesday, June 20, 2018
Information-Rich 13C Satellites
Seemingly simple NMR spectra often contain much more information than one might think. For example, the 1H NMR spectrum of 1,4-dioxane is primarily a singlet from which one obtains only an isotropic 1H chemical shift value. There is however much more information available in the spectrum which is often not recognized or used. The 1H NMR spectrum of a naturally occurring sample of 1,4-dioxane is the weighted sum of the 1H spectra of all possible isotopomers. It is the dominant tetra-12C isotopomer that gives rise to the singlet but since 13C (spin I = 1/2) is 1.1% naturally abundant, one expects to observe also the mono-13C isotopomer. The di-, tri- and tetra-13C isotopomers are very rare and can be neglected. The symmetry in the mono-13C isotopomer is lost compared to the tetra-12C isotopomer and one obtains a complex second-order spectrum, part of which can be represented by an AA'BB'X spin system. The spectrum of the AA'BB'X spin system depends on many more parameters than just the isotropic 1H chemical shift. This is illustrated in the figure below.

The bottom panel of the figure is the measured 300 MHz 1H NMR spectrum of 1,4-dioxane with an exaggerated vertical scale to accentuate the 13C satellites resulting from the protons color coded in pink in the mono-13C isotopomer. The large central region of the spectrum is the result of all the protons color coded in yellow from both the tetra-12C and mono-13C isotopomers. A simulation of this second-order spectrum was calculated from the parameters below and is shown in the top panel of the figure.
 Any isotope shifts in the 1H frequencies due to 13C vs 12C bonding were neglected in the simulation. The fit of the simulation to the 13C satellites is particularly sensitive to 1JC-Ha, 1JC-Hb, 3JHa-Hc, 3JHa-Hd, 3JHb-Hc and 3JHb-Hd and much less sensitive to 2JC-Hc, 2JC-Hd, 2JHa-Hb and 2JHc-Hd. A fit of the simulation to the experimental spectrum produces estimates for all of the coupling constants in the AA'BB'X spin system - much more information than a single 1H isotropic chemical shift!
Any isotope shifts in the 1H frequencies due to 13C vs 12C bonding were neglected in the simulation. The fit of the simulation to the 13C satellites is particularly sensitive to 1JC-Ha, 1JC-Hb, 3JHa-Hc, 3JHa-Hd, 3JHb-Hc and 3JHb-Hd and much less sensitive to 2JC-Hc, 2JC-Hd, 2JHa-Hb and 2JHc-Hd. A fit of the simulation to the experimental spectrum produces estimates for all of the coupling constants in the AA'BB'X spin system - much more information than a single 1H isotropic chemical shift!

The bottom panel of the figure is the measured 300 MHz 1H NMR spectrum of 1,4-dioxane with an exaggerated vertical scale to accentuate the 13C satellites resulting from the protons color coded in pink in the mono-13C isotopomer. The large central region of the spectrum is the result of all the protons color coded in yellow from both the tetra-12C and mono-13C isotopomers. A simulation of this second-order spectrum was calculated from the parameters below and is shown in the top panel of the figure.

Wednesday, June 13, 2018
Distortions due to Lock Saturation
The amplitude of the 2H lock signal provides information for an electronic feedback circuit which continuously corrects the magnetic field strength (by way of a B0 shim) to compensate for environmental instability. A poor 2H lock signal will provide unreliable input for the feedback circuit and B0 compensation will be erratic. This leads to undesirable effects in NMR spectra. For example, noisy lock signals will lead to undesirable noise at the base of the observed NMR peaks. If one uses too much lock power, the 2H lock signal gets saturated and the lock amplitude is unstable. A saturated 2H lock will lead to problems in the NMR spectrum since the input to the B0 compensation feedback circuit is unstable. This is demonstrated in the figure below.
 When one scan is collected, there are spectral distortions at the base of the NMR resonances. When 16 scans are collected these artifacts average to produce a general broadening at the base of the NMR resonances. Be careful not to saturate the 2H lock.
When one scan is collected, there are spectral distortions at the base of the NMR resonances. When 16 scans are collected these artifacts average to produce a general broadening at the base of the NMR resonances. Be careful not to saturate the 2H lock.

Friday, June 1, 2018
The limitations of 19F GARP Decoupling
In a previous post, it was shown that distorted line shapes are obtained for resonances in broadband decoupled NMR spectra when the resonances of the decoupled nuclide are outside of the effective decoupling bandwidth. This can be a particularly difficult problem when observing 1H NMR spectra with 19F decoupling. 19F has a large chemicals shift range so, if there are multiple widely spaced 19F resonances, it will be difficult or impossible to decouple all 19F sites at once, particularly at higher magnetic field strengths. If one is not aware of this problem, data misinterpretation may be an issue as distorted line shapes will lead incorrect splittings used to measure coupling constants. The figure below illustrates this problem. The top three panels of the figure show the 300 MHz 1H[19F] NMR spectra for the three 1H resonances of 1,2-difluoropyridine as a function of the 19F decoupler offset. The GARP decoupling scheme was used with 90° pulses of 80 µsec. The decoupler offsets, depicted in the bottom panel of the figure, were varied in 5 ppm increments.
 Of the 11 decoupler offsets used, only offset 6 (at -116 ppm) effectively decoupled both 19F sites. Varying the decoupler offset by only ± 5 ppm leads to distorted line shapes, which are particularly pronounced for the H3 resonance. These distorted line shapes could easily lead to data misinterpretation and erroneous coupling constants. In this case, the 19F decoupling bandwidth is 55 ppm. Since the chemical shift difference between the two 19F resonances is 52 ppm, one is able to obtain a fully 19F decoupled 1H spectrum with the careful choice of the decoupler offset frequency however, there will be cases where the decoupling bandwidth would not be sufficient to decouple all 19F resonances in some molecules. How then can one generally evaluate all of the coupling constants in fluorine containing molecules? The 19F-19F couplings can be evaluated in a 19F[1H] spectrum (not shown). Specific 1H-19F coupling constants can be determined by measuring a 1H PSYCHE spectrum or be collecting 1H spectra with selective 19F continuous wave (CW) decoupling for each of the19F resonances. The latter is shown in the figure below. The bottom panel shows a standard 1H spectrum. The middle two panels show the 1H spectra for each of the 19F sites decoupled separately using CW decoupling. The top panel shows the fully 19F decoupled spectrum.
Of the 11 decoupler offsets used, only offset 6 (at -116 ppm) effectively decoupled both 19F sites. Varying the decoupler offset by only ± 5 ppm leads to distorted line shapes, which are particularly pronounced for the H3 resonance. These distorted line shapes could easily lead to data misinterpretation and erroneous coupling constants. In this case, the 19F decoupling bandwidth is 55 ppm. Since the chemical shift difference between the two 19F resonances is 52 ppm, one is able to obtain a fully 19F decoupled 1H spectrum with the careful choice of the decoupler offset frequency however, there will be cases where the decoupling bandwidth would not be sufficient to decouple all 19F resonances in some molecules. How then can one generally evaluate all of the coupling constants in fluorine containing molecules? The 19F-19F couplings can be evaluated in a 19F[1H] spectrum (not shown). Specific 1H-19F coupling constants can be determined by measuring a 1H PSYCHE spectrum or be collecting 1H spectra with selective 19F continuous wave (CW) decoupling for each of the19F resonances. The latter is shown in the figure below. The bottom panel shows a standard 1H spectrum. The middle two panels show the 1H spectra for each of the 19F sites decoupled separately using CW decoupling. The top panel shows the fully 19F decoupled spectrum.
 Using these data, all of the coupling constants can be evaluated and are shown in the figure below.
Using these data, all of the coupling constants can be evaluated and are shown in the figure below.
 In conclusion, one must be careful in interpreting 1H[19F] spectra and understand the limits of the 19F decoupling scheme used.
In conclusion, one must be careful in interpreting 1H[19F] spectra and understand the limits of the 19F decoupling scheme used.



Tuesday, May 29, 2018
Decoupling Bandwidth and Distorted Line Shapes
Broadband X nucleus decoupling (X = 13C, 15N, 31P, 11B, 19F etc.....) is frequently used in 1H detected 2D HSQC/HMQC data collection or in standard 1D 1H spectra to aid in structure assignment. When broadband decoupling schemes are used, one must keep in mind that they are not infinitely broadbanded. They have finite bandwidths over which they are effective thus limiting the chemical shift range for the decoupled nuclide. The effective bandwidth depends on the particular decoupling scheme and the decoupling power used. If multiple peaks are to be decoupled, one must insure that all peaks are within the decoupler bandwidth. One can determine experimentally the effective decoupling bandwidth by running a series of 1H spectra varying the decoupler offset frequency. Such a measurement is shown in the figure below for the P-CH3 methyl resonance of dimethyl methylphosphonate. 300 MHz 1H [31P] NMR spectra were collected in a pseudo-2D fashion, incrementing the decoupler offset frequency from 200 ppm to -200 ppm from the 31P resonance frequency in 1 ppm steps. Broadband GARP decoupling was employed with a power of 3125 Hz (80 µsec 90° pulses). The pseudo-2D contour plot is shown in the left-hand panel and a stacked plot is shown in the right-hand panel.
 One can see that the effective decoupling bandwidth is 15.56 kHz or 128 ppm on a 300 MHz instrument. When the decoupler offset exceeds ±64 ppm from the 31P resonance frequency, one obtains distorted line shapes. Representative distorted line shapes are shown in the figure below. The bottom spectrum was collected with no 31P decoupling. The top, fully decoupled spectrum was collected with on-resonance 31P GARP decoupling. The middle spectra, highlighted in pink, are representative distorted spectra outside of the effective decoupling bandwidth.
One can see that the effective decoupling bandwidth is 15.56 kHz or 128 ppm on a 300 MHz instrument. When the decoupler offset exceeds ±64 ppm from the 31P resonance frequency, one obtains distorted line shapes. Representative distorted line shapes are shown in the figure below. The bottom spectrum was collected with no 31P decoupling. The top, fully decoupled spectrum was collected with on-resonance 31P GARP decoupling. The middle spectra, highlighted in pink, are representative distorted spectra outside of the effective decoupling bandwidth.
 If one is not aware of the decoupling offset and available bandwidth, one may obtain misleading line shapes subject to misinterpretation.
If one is not aware of the decoupling offset and available bandwidth, one may obtain misleading line shapes subject to misinterpretation.


Tuesday, May 15, 2018
NMR of Toothpaste
Some common household products contain many NMR active nuclides able to provide information on the identify the major components of the product. Toothpaste is such an example. It contains abrasives, surfactants, cleansers, fluoride, sweeteners, foaming agents, flavors, etc.... A survey of some of the NMR active nuclides can reveal the major components. The figure below shows the 19F, 31P, 23Na, 13C and 1H NMR spectra of a D2O slurry of Crest Complete toothpaste acquired on a 300 MHz spectrometer as well as the 29Si CP/MAS NMR spectrum of a sample of dried Crest Complete toothpaste, collected on a 200 MHz spectrometer. Except for the 29Si CP/MAS spectrum, which was collected over several hours, all other spectra were collected in a matter of minutes.
 The 19F spectrum is consistent with the fluoride ion which is a well known agent for preventing tooth decay. The 31P spectrum collected with 1H decoupling shows two major peaks consistent with diphosphate and phosphate anions. Salts of these anions are used as water retention agents, stabilizers and emulsifiers. The 23Na spectrum shows a single peak consistent with sodium cations, balancing the charge for the fluoride, diphosphate and phosphate anions. The 13C and 1H NMR spectra show one major component consistent with sorbitol, commonly used as a sweetener. Other minor components are evident in both the aliphatic and aromatic regions of the 1H and 13C spectra. The 29Si CP/MAS spectrum of the dried toothpaste is consistent with silica, which is used as an abrasive. The two peaks are due to Q4 (Si(OSi)4) and Q3 (Si(OH)(0Si)3) silicon sites. It should be noted that there are many other components including flavoring agents, coloring agents and preservatives present in concentrations which would require much more time and attention to identify.
The 19F spectrum is consistent with the fluoride ion which is a well known agent for preventing tooth decay. The 31P spectrum collected with 1H decoupling shows two major peaks consistent with diphosphate and phosphate anions. Salts of these anions are used as water retention agents, stabilizers and emulsifiers. The 23Na spectrum shows a single peak consistent with sodium cations, balancing the charge for the fluoride, diphosphate and phosphate anions. The 13C and 1H NMR spectra show one major component consistent with sorbitol, commonly used as a sweetener. Other minor components are evident in both the aliphatic and aromatic regions of the 1H and 13C spectra. The 29Si CP/MAS spectrum of the dried toothpaste is consistent with silica, which is used as an abrasive. The two peaks are due to Q4 (Si(OSi)4) and Q3 (Si(OH)(0Si)3) silicon sites. It should be noted that there are many other components including flavoring agents, coloring agents and preservatives present in concentrations which would require much more time and attention to identify.

Wednesday, March 28, 2018
Appropriate Choice of Presaturation Time
Presaturation is one of the most common methods of solvent suppression. A long selective low power pulse is applied at the solvent frequency followed by a hard non-selective read pulse (or composite pulse). Aside from a well-shimmed homogeneous magnet, there are two important parameters required for effective presaturation: saturation power and saturation time. The selection of saturation power was addressed in a previous post. With a properly selected saturation power, the appropriate choice for the saturation time depends on the relaxation properties of the solvent and the B1 field inhomogeneity of the probe. To determine an appropriate saturation time experimentally, one can run a pseudo 2D saturation pulse sequence like the one in the figure below.
 This sequence uses a recycle delay, D1, which is the sum of the incremented presaturation time and a resting delay. Each FID is Fourier transformed but no Fourier transform is done with respect to the incremented presaturation time. The result of this sequence for a plant extract dissolved in H2O/D2O on a 300 MHz spectrometer using a saturation power of 38.4 Hz (54 dB) and a recycle time, D1, of 5 seconds is shown in the figure below.
This sequence uses a recycle delay, D1, which is the sum of the incremented presaturation time and a resting delay. Each FID is Fourier transformed but no Fourier transform is done with respect to the incremented presaturation time. The result of this sequence for a plant extract dissolved in H2O/D2O on a 300 MHz spectrometer using a saturation power of 38.4 Hz (54 dB) and a recycle time, D1, of 5 seconds is shown in the figure below.
 A partial proton spectrum is displayed on the horizontal axis with the saturation time on the vertical axis, incremented in 50 msec steps. Several selected 1D spectra are shown on the right with the vertical scale adjusted such that the water signal at 4.7 ppm is at full-scale. Clearly, resonances of the plant extract at 5.08 ppm, 4.51 ppm and 4.48 ppm are independent of the saturation time, whereas the water signal decays as a function of saturation time. For saturation times between 0 and ~1.3 sec, the intensity of the water signal follows a decaying sinusoidal curve with positive or negative phases depending on the duration of the saturation pulse. For the saturation power used in this measurement, the 90° pulse is 6.5 msec therefore the trend observed is not the primary 1H nutation curve but results from sampling the primary nutation curve in 50 msec increments. Since the Nyquist sampling condition is not met with sampling intervals of 50 msec, one observes an aliased nutation curve with a much lower frequency. The overall decay is due to relaxation and B1 inhomogeneity. After ~1.3 seconds, the water signal is saturated and the data are invariant for longer presatutation times. These data suggest that the minimum saturation time should be set >1.3 sec.
A partial proton spectrum is displayed on the horizontal axis with the saturation time on the vertical axis, incremented in 50 msec steps. Several selected 1D spectra are shown on the right with the vertical scale adjusted such that the water signal at 4.7 ppm is at full-scale. Clearly, resonances of the plant extract at 5.08 ppm, 4.51 ppm and 4.48 ppm are independent of the saturation time, whereas the water signal decays as a function of saturation time. For saturation times between 0 and ~1.3 sec, the intensity of the water signal follows a decaying sinusoidal curve with positive or negative phases depending on the duration of the saturation pulse. For the saturation power used in this measurement, the 90° pulse is 6.5 msec therefore the trend observed is not the primary 1H nutation curve but results from sampling the primary nutation curve in 50 msec increments. Since the Nyquist sampling condition is not met with sampling intervals of 50 msec, one observes an aliased nutation curve with a much lower frequency. The overall decay is due to relaxation and B1 inhomogeneity. After ~1.3 seconds, the water signal is saturated and the data are invariant for longer presatutation times. These data suggest that the minimum saturation time should be set >1.3 sec.


Tuesday, March 20, 2018
Field Dependence of a Simple Spin System
With the recent re-emergence of low-field NMR spectrometers at proton frequencies of 40, 60, 80 and 100 MHz, many younger NMR users (who have grown up with high-field spectrometers) are encountering more and more second-order spectra. These spectra are observed when the frequency difference between signals is comparable to the coupling between them. On a 600 MHz spectrometer, 1 ppm in a 1H spectrum = 600 Hz while on a 60 MHz spectrometer, 1 ppm in a 1H spectrum is only 60 Hz. Unlike frequency differences between signals (in Hz) which depend on the field strength, the coupling between signals (in Hz) is field invariant. Easily interpreted first-order spectra on high-field instruments can be information rich but much more complicated second-order spectra on low-field instruments. The figure below shows simulated 1H NMR spectra of a fictitious isolated ethyl group as a function of field strength. The difference in chemical shift between the -CH3 and -CH2- signals is 0.5 ppm and the 3JH-H coupling constant is 10 Hz. The spectra are plotted on a ppm scale on the left and on a Hz scale on the right. At higher fields, one immediately recognizes the familiar triplet and quartet. At lower fields, the spectra are much more complicated. The signals are closer to one another (in Hz) and therefore have more second-order character as the frequency difference between signals becomes comparable to the coupling between them.


Thursday, February 8, 2018
How Much Presaturation Power is Needed?
Measuring 1H NMR spectra of samples in water (or mixtures of H2O and D2O) usually requires some form of solvent suppression of which selective presaturation is the most common method. In this technique, a long low power selective pulse is applied before the high power excitation pulse (or composite pulse). If too little power is used for presaturation, the water signal will not be sufficiently suppressed. If too much power is used, one loses intensity of signals close to the water resonance and foregoes the quantitative nature of the NMR data. The question then arises as to how much power is required for presaturation. The figure below shows the 300 MHz 1H NMR spectra of a plant extract in H2O/D2O measured as a function of presaturation power. The data were acquired on a Bruker AVANCE II console with the Bruker zgcppr pulse sequence using a two second presaturation pulse. The power levels are reported in dB and Hz.

One can see that at higher presaturation powers, one loses intensity for peaks near the water signal. In this case, where the nearest resonance of interest is 52 Hz from the water signal, one can obtain unattenuated signals with a presaturation field strength of 27 Hz. This corresponds to 58 dB on this instrument.
Thursday, January 4, 2018
Improved Solvent Suppression with Composite Pulses
A hard rf pulse delivers an rf field to the NMR sample inside of the coil. The rf field is not perfectly homogeneous nor does it end abruptly at the edge of the coil. When a long, selective, low power presaturation pulse is given to suppress a water signal, the water in the coil may be fully saturated whereas water outside of the coil will not be. Furthermore, unsaturated water outside of the coil may be outside of the carefully shimmed region of the magnet and give rise to a broadened residual signal when presaturation is used prior to a hard 90° excitation pulse. One long known* way to avoid this residual broad signal is to use a composite 90° pulse after presaturation. One of the simplest such pulses is a (90°x - 90°y - 90°-x - 90°-y) composite pulse which is designed to excite sample inside of the coil but not outside of the coil. As a result, when it is used after a water presaturation pulse, it will not excite the broad signal from the unsaturated water outside of the coil and it will provide a spectrum with better presaturation performance. The figure below shows a small portion of the 1H spectrum of a plant extract in H2O/D2O.
 The water signal is highlighted in pink. The left panel shows a conventional spectrum acquired with a 90° pulse. The center panel is a spectrum of the same sample where a two second low power presaturation pulse preceded the 90° hard pulse (Bruker pulse program = zgpr). One can see that most of the water is suppressed from the presaturation pulse however, a broad water signal remains from the water outside of the coil. The spectrum in the right-hand panel is the same as that in the center except that the 90° pulse was replaced with a composite 90° pulse (Bruker pulse program = zgcppr). Clearly, the broad signal from the unsaturated water outside of the coil is essentially gone providing a spectrum with much better water suppression.
The water signal is highlighted in pink. The left panel shows a conventional spectrum acquired with a 90° pulse. The center panel is a spectrum of the same sample where a two second low power presaturation pulse preceded the 90° hard pulse (Bruker pulse program = zgpr). One can see that most of the water is suppressed from the presaturation pulse however, a broad water signal remains from the water outside of the coil. The spectrum in the right-hand panel is the same as that in the center except that the 90° pulse was replaced with a composite 90° pulse (Bruker pulse program = zgcppr). Clearly, the broad signal from the unsaturated water outside of the coil is essentially gone providing a spectrum with much better water suppression.
*A. Bax. J. Magn. Res. 65, 142 (1985).

*A. Bax. J. Magn. Res. 65, 142 (1985).
Subscribe to:
Posts (Atom)
