 A better idea is to save spectrometer time, make the acquisition time shorter and avoid collecting the noise in the first place.
A better idea is to save spectrometer time, make the acquisition time shorter and avoid collecting the noise in the first place.
Friday, November 30, 2007
Throwing Away Noise to Improve Your Data
It is no surprise that the Fourier transform of noise is noise, so getting rid of noise in you FID will get rid of it from your spectrum as well. If your FID decays long before the acquisition time is over then you have collected unnecessary noise and your spectrum will have a lower than necessary signal-to-noise ratio. You can throw away the noise and replace it with null points before carrying out the Fourier transform. This is shown in the figure below where the improvement in the signal-to-noise ratio is obvious.A better idea is to save spectrometer time, make the acquisition time shorter and avoid collecting the noise in the first place.
A better idea is to save spectrometer time, make the acquisition time shorter and avoid collecting the noise in the first place.
Thursday, November 29, 2007
Nyquist Fold-backs in Magnetic Resonance Images
Nyquist fold-backs are not just limited to 1D and 2D NMR data. They can also be observed in magnetic resonance images. The figures below are magnetic resonance images of seedless grapes acquired on our AVANCE 500. The Nyquist fold-backs are circled in blue. 
Tuesday, November 27, 2007
How Fast Should I Spin My Solid Sample?
The choice of MAS rate depends on the interaction to be averaged by the magic angle spinning. If you want to average out the chemical shielding anisotropy then you must spin the sample at a rate comparable to or greater than the span of the chemical shift tensor expressed in Hz. Although the spans of chemical shift tensors, measured in ppm, are independent of field strength they are linearly dependent on field strength, when expressed in Hz. As a result, an appropriate spinning speed in one magnet may not be an appropriate spinning speed for the same sample in another magnet. The spectra below illustrate this point. Both are 31P CPMAS spectra of dibasic ammonium phosphate with a spinning speed of 4 kHz. The lower trace was acquired at 11,75 Tesla while the upper trace was collected at 4.7 Tesla. One can see that there are many more spinning sidebands in the spectrum acquired at higher field despite the identical spinning speeds. One would have to spin the sample at 10 kHz in an 11.75 Tesla magnet to get a spectrum comparable to the one acquired with a spinning speed of 4 kHz in a 4.7 Tesla magnet.
One would have to spin the sample at 10 kHz in an 11.75 Tesla magnet to get a spectrum comparable to the one acquired with a spinning speed of 4 kHz in a 4.7 Tesla magnet.
One would have to spin the sample at 10 kHz in an 11.75 Tesla magnet to get a spectrum comparable to the one acquired with a spinning speed of 4 kHz in a 4.7 Tesla magnet.
Monday, November 26, 2007
HMQC Responses in HMBC Data
Despite the built in low pass J filters written into the HMBC pulse sequences, the much larger one-bond 1H - 13C coupling responses are not fully suppressed. These one-bond HMQC responses show up as a pair of responses in the acquisition domain separated by the one-bond 1H - 13C coupling. The responses are doubled since 13C decoupling is not typically used in HMBC sequences. An example of these artifacts is shown below.
Friday, November 23, 2007
31P Decoupling
All of you routinely use proton decoupling in your 31P NMR spectra, many of you without giving it a thought. Few people realize that it is a simple matter to decouple 31P in your 1H NMR spectra. This will allow you to determine which protons are coupled to phosphorus. An example of this is shown below. The lower trace is a standard 1H spectrum and the upper trace is a 1H spectrum with 31P decoupling.

Thursday, November 22, 2007
Baseline Correction in Satellite Transition MAS Spectra of Quadrupolar Nuclei
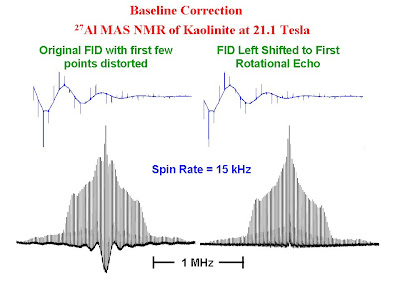
When one acquires NMR data with extremely short dwell times (or very large spectral widths), problems are often encountered with severe baseline roll as the first few points in the FID are lost either to the receiver dead time or probe ringing. The satellite transition MAS spectra of quadrupolar nuclei are characterized by many sharp spinning sidebands and require the use of very large spectral widths to accommodate the entire sideband envelope. The FIDs for such samples contain rotational echoes at the period of the rotor. Due to the extremely short dwell times required to collect such data, baseline problems are very common. In such cases simply removing the initial few bad points or using backward linear prediction will not solve the problem satisfactorily due to the nature of the FID. There is however a very simple solution. One can simply remove all of the points in the FID before the first rotational echo and then carry out the Fourier transform. The figure below shows the FID's and spectra for the 27Al MAS NMR of kaolinite. The spectra show the satellite transitions. The resonance for the central transition is off scale in the figure. In the left hand side of the figure the raw FID was Fourier transformed and phase corrected. One can see extreme baseline roll. In the right hand side of the figure, all of the points prior to the center of the first rotational echo in the FID were discarded and the new FID was Fourier transformed and phase corrected. The baseline is much improved.
Wednesday, November 21, 2007
Magic Angle Spinning
The NMR spectra of solids generally have very broad lines due to a number of interactions which are averaged either to zero or an isotropic value in solution. It is the averaging of these interactions which leads to the narrow lines observed in liquids. The rapid random motion responsible for the averaging in solution is not present for solids, however, it can be mimicked by spinning the sample about an angle of 54.7 degrees with respect to the magnetic field at a rate comparable to or greater than the extent of the interaction being averaged. This technique is called magic angle spinning (MAS) and it allows the measurement of high resolution spectra of solids. When the spinning rate is less than the extent of the interaction, the spectrum is split into sidebands spaced at the spinning speed. In the figure below are 81 MHz 31P CPMAS spectra of ammonium dihydrogen phosphate with high power proton decoupling at various spinning rates. When the spinning speed is zero, one can see the powder spectrum characterizing the chemical shift interaction. As the spinning speed is increased, there is a centerband at the isotropic chemical shift with sidebands whose intensities can be used to calculate the chemical shift tensor. When the spinning speed exceeds the span of the chemical shift interaction the sidebands are very small and a liquid-like spectrum is obtained. 
Tuesday, November 20, 2007
Excitation Profiles
The range of frequency over which NMR resonances are excited depends on the duration of the monochromatic radio frequency pulse applied to the sample. Very long pulses will excite a very narrow range of frequencies whereas very short pulses will excite very wide frequency ranges. For example, presaturation pulses used for solvent suppression, are typically several seconds long while non-selective pulses are typically micro seconds in duration. It is essential to use very short pulses to provide even excitation over large spectral widths. The shape of the excitation profile is related to the Fourier transform of the pulse.

Monday, November 19, 2007
Increasing the Signal-to-Noise Ratio in Solids MAS Spectra
Often MAS or CPMAS NMR spectra of solids will have several spinning sidebands. In such spectra, where there are no other complications, one can increase the signal-to-noise ratio for the isotropic signals by simply adding the sideband intensity to the isotropic spectrum. Below is a 50.68 MHz, room temperature, 15N CPMAS spectrum of a clay sample which absorbed some 15N labelled pyridine and then was heated to 400 degrees. The spinning speed was set to 2.5 kHz. The full spectrum is shown in the bottom panel. The isotropic region is shown on the top left panel. The spectrum in the top right panel was obtained by shifting the spectrum by multiples of the spinning speed and adding the shifted spectra to the original spectrum. Note that the isotropic region of the spectrum has a much improved signal-to-noise ratio.

Friday, November 16, 2007
DEPT and DEPTQ
Many of you run both standard 13C and 13C DEPT-135 spectra of your compounds. This may take a considerable period of time (and cost a considerable amount of money) if your sample is dilute. You can run a single DEPTQ-135 spectrum of your compound and get the same amount of information. This may save a great deal of spectrometer time. A DEPTQ-135 spectrum is the same as a DEPT-135 spectrum except the quaternary carbons are present and 180 degrees out of phase with respect to the CH and CH3 carbons. Note also that the commonly used deuterated solvents will show up in a DEPTQ spectrum and can be used for chemical shift referencing. Below is the standard 13C spectrum, the DEPT-135 spectrum and the DEPTQ-135 spectrum of ethylbenzene.
Thursday, November 15, 2007
Zero Filling
Zero filling is a data processing technique where zero points are appended to the free induction decay before Fourier transformation. The effect of zero filling is to increase the digital resolution in the spectrum. Since the zeros contain no new information, there is no new information added to the spectrum. You can think of it as artificially increasing the acquisition time after the data collection without adding any new information or true resolution to the spectrum.
 To zero fill on a Bruker spectrometer, the "si" parameter is increased to a value greater than the number of points collected in the free induction decay. On a Varian spectrometer, the corresponding parameter is "fn".
To zero fill on a Bruker spectrometer, the "si" parameter is increased to a value greater than the number of points collected in the free induction decay. On a Varian spectrometer, the corresponding parameter is "fn".
To zero fill on a Bruker spectrometer, the "si" parameter is increased to a value greater than the number of points collected in the free induction decay. On a Varian spectrometer, the corresponding parameter is "fn".
Wednesday, November 14, 2007
HSQC and Edited HSQC Spectra
Many of you use a simple magnitude HMQC sequence to establish heteronuclear one-bond 1H -13C correlations. One can also use the phase sensitive HSQC sequences to obtain the same information. Although the data must be phased by the user, the phase sensitive sequences will provide data with higher resolution as absorption line shapes are much narrower than the magnitude signals obtained in non-phase-sensitive sequences. There is also an edited HSQC sequence available which provides multiplicity information similar to that of a 13C DEPT-135 sequence where CH and CH3 signals are phased up and CH2 signals are phased down. In the figure below is the HSQC and edited HSQC spectra of 3-heptanone. The red CH2 signals in the edited HSQC are negative with respect to the black CH3 signals. Using this sequence may save you time (and money) as there will be no need to run a 13C DEPT spectrum.

Tuesday, November 13, 2007
What Mixing Time Should I Use for My 2D-NOESY Measurements?
NOEs are often very small and the appropriate choice of mixing time is a critical factor in achieving good NOESY spectra. There are two opposing factors which must be considered when choosing the most appropriate mixing time. Firstly, you would like the NOE to build up for as long as possible during the mixing time and secondly, you want to loose as little signal as possible to relaxation during the mixing time. The best compromise is to choose a mixing time equal to the average T1 relaxation times for the signals in which you are interested. (See the BLOG entry for October 31, 2007, to learn how to estimate T1's.)
 If the standard pulse programs are used, the mixing time is the "d8" parameter on a Bruker spectrometer and the "mix" parameter on a Varian spectrometer.
If the standard pulse programs are used, the mixing time is the "d8" parameter on a Bruker spectrometer and the "mix" parameter on a Varian spectrometer.
If the standard pulse programs are used, the mixing time is the "d8" parameter on a Bruker spectrometer and the "mix" parameter on a Varian spectrometer.
Monday, November 12, 2007
Testing a Magnet for Field Drift
One can test a magnet for drift by measuring a spectrum unlocked with multiple scans using a very long recycle delay. The entire spectrum will shift between scans according to the magnet drift, therefore you should have as many multiples of the spectrum as the number of scans. In the figure below, the drift rate for a 500 MHz wide bore magnet was measured. A 16 scan 1H spectrum was acquired for CHCl3 in acetone-d6 without locking. The recycle delay was 1 hour. The resonance shifts to lower frequency as a function of time. One can also see that the shim currents (either the cryoshims or room temperature shims) have drifted over the course of the measurement accounting for the broadening and multiplicity at later times.

Sunday, November 11, 2007
Learn How to Play Music with Your NMR Spectrometer
I always thought this site by Walter Bauer was a lot of fun. The difference frequency between an NMR resonance and the carrier frequency is used creatively to make music. Listen to your spectrometer!
http://www.chemie.uni-erlangen.de/oc/research/NMR/music.html
NMR spectrometers are really just very expensive musical instruments!
http://www.chemie.uni-erlangen.de/oc/research/NMR/music.html
NMR spectrometers are really just very expensive musical instruments!
Friday, November 9, 2007
Increasing the Signal-to-Noise Ratio in Solid State Wideline 2H Spectra
At low magnetic field strengths where chemical shielding anisotropy is not a problem, the wide line 2H spectra of solids produce symmetrical spectra. One can increase the signal-to-noise ratio in these spectra by a factor of the square root of two by reversing the spectrum and adding it to itself as illustrated below for the 2H spectrum of perdeuterated poly-methyl methacrylate.

Thursday, November 8, 2007
Why Does My Quaternary Alkyne Carbon Show Up in My 13C DEPT Spectrum?
The BLOG entry for September 20, 2007 illustrated how some signals from protonated carbons can be either missing or of much reduced intensity in 13C DEPT spectra. This is due to unusual one-bond 13C-1H coupling constants. There are also cases where quaternary carbon signals will show up in 13C DEPT spectra due to unusually large two-bond 13C-1H coupling constants. Such is the case for the alkyne in the figure below. A standard 13C DEPT-135 sequence optomized for a 13C-1H coupling of 145 Hz (the average one-bond 13C-1H coupling constant) was acquired and compared to the standard 13C spectrum with 1H decoupling. One can see that the protonated alkyne carbon has less intensity than expected as its one-bond 13C-1H coupling constant is ~ 250 Hz. Also the quaternary alkyne carbon is present in the spectrum as its 2-bond 13C-1H coupling constant is ~ 50 Hz. Although neither of these couplings is close to 145 Hz, they are both of a magnitude where one would expect to see a signal in the DEPT 135 spectrum.

Wednesday, November 7, 2007
Getting Incorrect Lineshape Specifications
One can easily fool themselves by getting line shape specifications (see BLOG entry for Tuesday October 23, 2007) that are better than the truth. Erroneous specifications will be obtained on the standard line shape sample (1% CHCl3 in acetone-d6) if sufficient time is not allowed for complete relaxation to occur and the height of the 13C satellites is used as a guide to measure the line width. This error results because the T1 of the 13CHCl3 isotopomer is shorter than that of the 12CHCl3 isotopomer. Therefore, if the signals are partially saturated, the 13C satellites will be over-represented with respect to the singlet. If the height of the satellites is used as a guide to measure the line width on the singlet, the line shape specifications will be narrower than they would be if complete relaxation was allowed to occur. This is illustrated in the figure below. The trace on the left is a fully relaxed single scan spectrum using a 90 degree pulse with an 8 second acquisition time. The trace on the right was acquired under identical conditions however the spectrum was partially saturated by collecting 15 dummy scans with an inter-pulse spacing of 8 seconds immediately prior the receiver being turned on. The spectra were scaled such that the singlets had the same height. Note the difference in signal-to-noise ratio and the relative heights of the satellites compared to the singlet.
 One can still obtain the correct line shape specification in a partially saturated spectrum by measuring the linewidths at 0.55% and 0.11% of the height of the singlet if the satellites are not used as a guide.
One can still obtain the correct line shape specification in a partially saturated spectrum by measuring the linewidths at 0.55% and 0.11% of the height of the singlet if the satellites are not used as a guide.
One can still obtain the correct line shape specification in a partially saturated spectrum by measuring the linewidths at 0.55% and 0.11% of the height of the singlet if the satellites are not used as a guide.Tuesday, November 6, 2007
19F - 1H HOESY Experiment
Many of you use proton NOESY spectra to establish whether or not two types of protons are close to one another in space. The same type of experiment is also possible between nuclei of different isotopes. This technique is called HOESY (Heteronuclear Overhauser Effect SpectroscopY) and can be used to assess the spatial proximity between 2 heteronuclei. Below is an example of a 19F detected 19F - 1H HOESY spectrum of a sugar. (sample courtesy of Jennifer Chaytor)

Monday, November 5, 2007
How Many Scans Should I Collect?
Many students ask this question. You should collect enough scans until the signal-to-noise ratio is adequate for your needs. For NMR measurements (where saturation is not a problem) the signal -to-noise ratio increases as the square root of the number of scans. In order to double the signal-to-noise ratio in a particular spectrum one must collect 4 times as many scans as were collected in the original spectrum. Take a look at the figure below where spectra are plotted on an absolute intensity scale against the number of scans collected.
 If the signal-to-noise ratio is only 2 after 1 hour, it will be < 7 after 12 hours and only 16 after 64 hours. If you do not see a signal in 1 hour it is probably not worth running the sample overnight!
If the signal-to-noise ratio is only 2 after 1 hour, it will be < 7 after 12 hours and only 16 after 64 hours. If you do not see a signal in 1 hour it is probably not worth running the sample overnight!
If the signal-to-noise ratio is only 2 after 1 hour, it will be < 7 after 12 hours and only 16 after 64 hours. If you do not see a signal in 1 hour it is probably not worth running the sample overnight!
Friday, November 2, 2007
FID Truncation and Spectral Distortion
Have you ever seen a spectrum like the one in the lower trace of the figure below?
 This sin(x)/x distortion at the base of an NMR line is the result of truncation of the FID. The acquisition time was not long enough to capture the entire time domain signal. In addition to the distortion at the base of the lines, there is also a loss in spectral resolution as "sharp" features in a spectrum are defined by later times in the free induction decays. If you see this, you should re-run your spectrum with a longer acquisition time. If you do not have the option of re-running the spectrum, increase the line broadening (LB) and re-transform the data. This will get rid of the distortion but will not help with the resolution.
This sin(x)/x distortion at the base of an NMR line is the result of truncation of the FID. The acquisition time was not long enough to capture the entire time domain signal. In addition to the distortion at the base of the lines, there is also a loss in spectral resolution as "sharp" features in a spectrum are defined by later times in the free induction decays. If you see this, you should re-run your spectrum with a longer acquisition time. If you do not have the option of re-running the spectrum, increase the line broadening (LB) and re-transform the data. This will get rid of the distortion but will not help with the resolution.
This sin(x)/x distortion at the base of an NMR line is the result of truncation of the FID. The acquisition time was not long enough to capture the entire time domain signal. In addition to the distortion at the base of the lines, there is also a loss in spectral resolution as "sharp" features in a spectrum are defined by later times in the free induction decays. If you see this, you should re-run your spectrum with a longer acquisition time. If you do not have the option of re-running the spectrum, increase the line broadening (LB) and re-transform the data. This will get rid of the distortion but will not help with the resolution.
Thursday, November 1, 2007
NMR to Distinguish Solid Polymorphs
Some solids have a number of different polymorphs (or phases). Being able to distinguish different polymorphs is very important for the pharmaceutical industry. Solid State NMR is able to distinguish between them. In the figure below is the 13C CPMAS spectrum of the solid amino acid, methionine acquired on a Bruker AVANCE 500 NMR spectrometer. The spectrum clearly shows two signals for each type of carbon.
 When the temperature is varied the intensity ratio of the peaks changes as one polymorph becomes more favored than the other.
When the temperature is varied the intensity ratio of the peaks changes as one polymorph becomes more favored than the other.
When the temperature is varied the intensity ratio of the peaks changes as one polymorph becomes more favored than the other.
Subscribe to:
Posts (Atom)
