It has always puzzled me how students so willingly accept the fact that
13C is J coupled to a spin
I = 1 nucleus like
2H (eg. the multiplets for the
13C spectra of deuterated solvents) however they do not question the fact that
13C almost never shows a J coupling to
14N (another spin
I = 1 nucleus). The reason that
13C exhibits J coupling to
2H is that the relaxation among the three energy levels of
2H is rather slow and each
13C "sees" the
2H in each of its three Zeeman states, splitting the
13C resonance into 3 lines of equal intensity. The efficiency of the relaxation among the energy levels of quadrupolar nuclei (among other things) depends on the magnitude of the quadrupolar coupling constant - the larger the quadrupolar coupling constant, the faster the relaxation. Unlike
2H, which has a very small quadrupolar coupling constant,
14N (and most other quadrupolar nuclei) has a substantial quadrupolar coupling constant and therefore the relaxation among its energy levels is very fast. The
13C therefore "sees" the
14N in a single "average" state and as a result is a singlet. There are however compounds where the
14N is in a very symmetric environment. The high symmetry make the quadrupolar coupling constant much smaller and the relaxation therefore much slower. In these cases one can observe the
13C -
14N J coupling. Depending on the relaxation of the
14N, the
13C lines can vary from being a sharp 1:1:1 triplet, to a broad unresolved triplet, to a very sharp singlet. Below is an example of a case where the
14N is in a very symmetric environment and the
13C -
14N J couplings are resolved.






 These are often called "soft" pulses.
These are often called "soft" pulses.


 A better idea is to save spectrometer time, make the acquisition time shorter and avoid collecting the noise in the first place.
A better idea is to save spectrometer time, make the acquisition time shorter and avoid collecting the noise in the first place.

 One would have to spin the sample at 10 kHz in an 11.75 Tesla magnet to get a spectrum comparable to the one acquired with a spinning speed of 4 kHz in a 4.7 Tesla magnet.
One would have to spin the sample at 10 kHz in an 11.75 Tesla magnet to get a spectrum comparable to the one acquired with a spinning speed of 4 kHz in a 4.7 Tesla magnet.


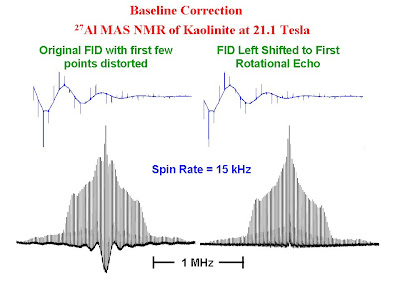




 To zero fill on a Bruker spectrometer, the "si" parameter is increased to a value greater than the number of points collected in the free induction decay. On a Varian spectrometer, the corresponding parameter is "fn".
To zero fill on a Bruker spectrometer, the "si" parameter is increased to a value greater than the number of points collected in the free induction decay. On a Varian spectrometer, the corresponding parameter is "fn".

 If the standard pulse programs are used, the mixing time is the "d8" parameter on a Bruker spectrometer and the "mix" parameter on a Varian spectrometer.
If the standard pulse programs are used, the mixing time is the "d8" parameter on a Bruker spectrometer and the "mix" parameter on a Varian spectrometer.



 One can still obtain the correct line shape specification in a partially saturated spectrum by measuring the linewidths at 0.55% and 0.11% of the height of the singlet if the satellites are not used as a guide.
One can still obtain the correct line shape specification in a partially saturated spectrum by measuring the linewidths at 0.55% and 0.11% of the height of the singlet if the satellites are not used as a guide.
 If the signal-to-noise ratio is only 2 after 1 hour, it will be < 7 after 12 hours and only 16 after 64 hours. If you do not see a signal in 1 hour it is probably not worth running the sample overnight!
If the signal-to-noise ratio is only 2 after 1 hour, it will be < 7 after 12 hours and only 16 after 64 hours. If you do not see a signal in 1 hour it is probably not worth running the sample overnight!
 This sin(x)/x distortion at the base of an NMR line is the result of truncation of the FID. The acquisition time was not long enough to capture the entire time domain signal. In addition to the distortion at the base of the lines, there is also a loss in spectral resolution as "sharp" features in a spectrum are defined by later times in the free induction decays. If you see this, you should re-run your spectrum with a longer acquisition time. If you do not have the option of re-running the spectrum, increase the line broadening (LB) and re-transform the data. This will get rid of the distortion but will not help with the resolution.
This sin(x)/x distortion at the base of an NMR line is the result of truncation of the FID. The acquisition time was not long enough to capture the entire time domain signal. In addition to the distortion at the base of the lines, there is also a loss in spectral resolution as "sharp" features in a spectrum are defined by later times in the free induction decays. If you see this, you should re-run your spectrum with a longer acquisition time. If you do not have the option of re-running the spectrum, increase the line broadening (LB) and re-transform the data. This will get rid of the distortion but will not help with the resolution.
 When the temperature is varied the intensity ratio of the peaks changes as one polymorph becomes more favored than the other.
When the temperature is varied the intensity ratio of the peaks changes as one polymorph becomes more favored than the other.
 These measurements can be very time consuming. One can get a reasonable estimate of the T1 much more quickly. Follow these simple steps:
These measurements can be very time consuming. One can get a reasonable estimate of the T1 much more quickly. Follow these simple steps: These spectra were acquired while the magnetic field was sweeping. This will happen on a Bruker spectrometer if you do not bother locking the field (or fixing the field when running unlocked). On rare occasions in automation, if there is difficulty in locking your sample, I have seem the spectrometer run a spectrum while sweeping the field. This however is very infrequent.
These spectra were acquired while the magnetic field was sweeping. This will happen on a Bruker spectrometer if you do not bother locking the field (or fixing the field when running unlocked). On rare occasions in automation, if there is difficulty in locking your sample, I have seem the spectrometer run a spectrum while sweeping the field. This however is very infrequent.
 Quadrature images are small reflections of large signals in the spectrum about the center. They are much less of a problem with newer instruments than they were with older ones. They become smaller as more scans are collected due to receiver phase cycling. You should be aware of quadrature images if you are searching for very small signals in the presence of very large ones.
Quadrature images are small reflections of large signals in the spectrum about the center. They are much less of a problem with newer instruments than they were with older ones. They become smaller as more scans are collected due to receiver phase cycling. You should be aware of quadrature images if you are searching for very small signals in the presence of very large ones.





 One should use this method with care as artificial cross peaks will appear for uncoupled signals with excessive t1 noise. Before symmetrization, one should look for the smallest real off-diagonal signal. Make a mental note of the signal. Symmetrize the spectrum and then scale it such that the smallest real off-diagonal signal noted above is the smallest signal in the symmetrized spectrum.
One should use this method with care as artificial cross peaks will appear for uncoupled signals with excessive t1 noise. Before symmetrization, one should look for the smallest real off-diagonal signal. Make a mental note of the signal. Symmetrize the spectrum and then scale it such that the smallest real off-diagonal signal noted above is the smallest signal in the symmetrized spectrum. 1. Use a standard setup and get more sample or collect more scans. Yeah yeah..... I know ..... if you could do this you wouldn't have a problem.
1. Use a standard setup and get more sample or collect more scans. Yeah yeah..... I know ..... if you could do this you wouldn't have a problem. Remember to tune the proton channel!
Remember to tune the proton channel!

 Many spectrometers will calculate the receiver gain automatically however you should be aware that this automatic calculation is not always perfect and that the receiver gain may have to be set manually. On a Varian spectrometer the receiver gain is the "gain" parameter. On a Bruker spectrometer the parameter is "rg". In both cases higher numbers mean a higher receiver gain.
Many spectrometers will calculate the receiver gain automatically however you should be aware that this automatic calculation is not always perfect and that the receiver gain may have to be set manually. On a Varian spectrometer the receiver gain is the "gain" parameter. On a Bruker spectrometer the parameter is "rg". In both cases higher numbers mean a higher receiver gain.
 If you inadvertently collect a spectrum with a Nyquist fold-back you can still calculate the correct chemical shift, as the signal will be the same number of ppm away from the wrong end of the axis as it is outside of the correct end of the axis.
If you inadvertently collect a spectrum with a Nyquist fold-back you can still calculate the correct chemical shift, as the signal will be the same number of ppm away from the wrong end of the axis as it is outside of the correct end of the axis.





