
Wednesday, April 30, 2008
23Na Background in NMR Tubes.
When we measure NMR spectra, we want to observe the NMR spectrum of our sample only. However, NMR probes, tubes (or MAS rotors) must be made of real materials, so we should expect some NMR background signals for the materials present inside (or near) the coil. The user should be aware of the presence of these signals. Instrument companies expend a great deal of effort to minimize the number and intensity of background signals in their NMR probes (this is one of the reasons they are so expensive). The figure below shows three 23Na NMR spectra taken on a high resolution NMR spectrometer. The top trace is the spectrum of an aqueous solution of NaCl, the middle trace is that of an empty NMR tube and the lower trace is that of an empty NMR probe. One can see that although the NMR probe is free of a 23Na background signal, the NMR tube is not.
Tuesday, April 29, 2008
1H - 11B HMQC
HMQC and HSQC experiments are very commonly applied to 1H - 13C and 1H - 15N. These techniques can also be applied to other isotopes. The figure below shows the 1H - 11B HMQC spectrum or ortho-carborane. The top and side traces are separately acquired 1H [11B] and 11B [1H] spectra, respectively. These data, combined with the 11B COSY data, allow the complete assignment of the 1H [11B] spectrum. 
Thank you to Dominique Duguay for providing the sample of ortho-carborane.
Thank you to Dominique Duguay for providing the sample of ortho-carborane.
Monday, April 28, 2008
11B COSY
Many NMR users are very familiar with the 1H COSY experiment. It is used extensively by organic chemists. Many people do not realize that the COSY method can be used for many other abundant isotopes as well. The figure below shows the 11B COSY spectrum of ortho-carborane. The data were collected with 1H decoupling and allow the complete assignment of the 11B NMR spectrum. Note that in this case, the 11B - 11B J couplings are unresolved in the 1D spectrum.  Thank you to Dominique Duguay for providing the sample for the figure.
Thank you to Dominique Duguay for providing the sample for the figure.
Thank you to Dominique Duguay for providing the sample for the figure.
Friday, April 25, 2008
New NMR BLOG for Structure Elucidation
Arvin Moser, an M.Sc. Chemistry graduate from the laboratory of Christian Detellier at the University of Ottawa and a current employee of the Advanced Chemistry Development (ACD) software company, has recently created a new BLOG called Philosophy to Chemistry to Elucidation. One of Arvin's responsibilities at ACD is to address the problems encountered by users of the software. In this capacity, he has insight into the questions and interests a large group of chemists. His BLOG addresses many of these questions. I think it is a worthwhile resource, especially for junior organic graduate students. Arvin's BLOG is now linked to this site. Click here to check it out.
This picture of a happy Arvin collecting a 23Na NMR spectrum was taken the day before he greased the door knob of my office. Oh yes, Arvin! I have not forgotten!
This picture of a happy Arvin collecting a 23Na NMR spectrum was taken the day before he greased the door knob of my office. Oh yes, Arvin! I have not forgotten!
Thursday, April 24, 2008
17O NMR
17O is a spin I = 5/2 quadrupolar nucleus with a natural abundance of only 0.037%. Its receptivity with respect to 13C is 0.061. Unlike the complications of the NMR spectra of quadrupolar nuclei in the solid state, the spectra in solution are quite simple as all of the energy levels are equally spaced by the Zeeman interaction, the first and second order quadrupolar effects averaged by the rapid isotropic motion in solution. 17O has a chemical shift range of more than 1600 ppm, making it quite sensitive to the local chemical environment of the oxygen and therefore potentially useful to the chemist. The figure below is a 17O NMR spectrum of a mixture of water, acetone, ethanol, isopropyl alcohol and ethyl acetate acquired in less than 15 minutes on a 300 MHz NMR spectrometer using a standard broadband probe with a 5 mm NMR tube. One can see that every single oxygen site is resolved in the spectrum.  In order to overcome the problem of low sensitivity, one should use as much sample as possible either as a neat liquid (if the sample is a liquid) or in a very concentrated solution with a solvent free of oxygen (if the sample is a solid) - the more sample the better. There may also be problems if the quadrupolar dominated relaxation rate is too fast resulting in resonances that are very broad.
In order to overcome the problem of low sensitivity, one should use as much sample as possible either as a neat liquid (if the sample is a liquid) or in a very concentrated solution with a solvent free of oxygen (if the sample is a solid) - the more sample the better. There may also be problems if the quadrupolar dominated relaxation rate is too fast resulting in resonances that are very broad.
In order to overcome the problem of low sensitivity, one should use as much sample as possible either as a neat liquid (if the sample is a liquid) or in a very concentrated solution with a solvent free of oxygen (if the sample is a solid) - the more sample the better. There may also be problems if the quadrupolar dominated relaxation rate is too fast resulting in resonances that are very broad.
Wednesday, April 23, 2008
Satellite Transitions in the Solid State NMR Spectra of Spin I= 5/2 Nuclei
The solid state MAS NMR spectra of spin I = n/2 (n = 1, 3, 5 ....) quadrupolar nuclei are more complicated than many people realize. There are chemical shielding anisotropy effects and both first order and second order quadrupolar effects which must be taken into account. Magic angle spinning averages out the first order quadrupolar effects and the chemical shielding anisotropy however, these interactions may show up as spinning sidebands. The field dependant second order quadrupolar effects are only partially averaged by magic angle spinning. In an MAS spectrum, each transition of the spin I = n/2 nucleus will have both a frequency shift and line shape characterized by the partially averaged second order quadrupolar interaction. The frequency shifts and lineshapes can provide valuable information about the quadrupolar coupling constant and the asymmetry parameter. The figure below shows the centerband region of the 27Al (I = 5/2) MAS NMR spectrum of aluminum-tris-acetylacetonate (Al(acac)3) acquired at 11.75 Tesla. There is only one aluminium site in the asymmetric unit of this compound. The major component of the spectrum is the central transition (m=1/2 - m= -1/2). It is the most intense as it is unaffected by the first order quadrupolar interaction and therefore does not have its intensity spread over multiple spinning sidebands. The first satellite transitions (m=3/2 - m= 1/2 and m= -1/2 - m= -3/2) are affected equally by the second order quadrupolar interaction and appear as a narrow resonance to higher frequency than the central transition. The second satellite transitions (m=5/2 - m= 3/2 and m= -3/2 - m= -5/2) are also affected equally by the second order quadrupolar interaction and appear as a weak broad resonance to higher frequency than both the first satellite transition and the central transition. Both satellite transitions are weak as their intensity is spread over many spinning sidebands (not shown). The second satellite transition is weaker than the first as it has a much broader line shape. 
Tuesday, April 22, 2008
Better 2D Data - More Scans or More Slices?
The question frequently arises: is it better to take more scans per increment of more increments in order to get higher quality 2D NMR data? In a 1D NMR spectrum acquired with sufficient relaxation, the signal-to-noise ratio increases with the square root of the number of scans taken. We can expect the same to be true of a 2D spectrum as well, therefore in order to double the signal-to-noise ratio one must collect 4 times as many scans per increment resulting in a four-fold increase in experiment time. Providing that there is more signal to be attained at longer evolution times, increasing the number of slices in a 2D data set increases the resolution in the F1 domain in a completely analogous way to increasing the acquisition time in a 1D data set. The higher F1 resolution implies narrower resonances and if the signal-to-noise ratio is measured based on peak height, then the overall signal-to-noise ratio is improved (as the area of the resonance must be constant). Also, the amount of overall signal in the 2D matrix is increased with the number of slices collected and therefore the overall signal-to-noise ratio is increased even more. Of course, if there is no more signal to be obtained at longer evolution times, then collecting more slices will only increase the amount of noise. HMQC data for 2-bromobutane is presented in the figure below showing the effects of increasing the number of scans vs the number if slices in such a way as to increase the experiment time by a factor of four. All of the data were scaled to have the largest signal at constant height. In this particular example where more signal was available at longer evolution times, the most improvement in the data is achieved by increasing the number of increments by a factor of four. 
It is worth noting that forward linear prediction in the F1 domain can also be used to increase the amount of signal and resolution while still maintaining short experiment times.
It is worth noting that forward linear prediction in the F1 domain can also be used to increase the amount of signal and resolution while still maintaining short experiment times.
Monday, April 21, 2008
How High Should the 2H Lock Signal Be?
New users of NMR spectrometers will often ask: How high should the 2H lock signal be? This question is asked while they are shimming the magnet. They want to know if they have "shimmed enough". There isn't really an answer to the question. The height of the lock signal depends on all of the following: which solvent is used, the amount of solvent in the NMR tube, the lock power, the lock gain, the lock phase and the magnet homogeneity. The lock signal is high enough when any further shimming cannot improve on the height.
Friday, April 18, 2008
The 13C and 13C DEPT Spectrum of "Acetone-d6"
A student recently asked me why her solvent resonance for acetone-d6 was showing up in her 13C DEPT spectrum. Since the methyls of acetone-d6 have no protons they will not show up in a DEPT spectrum however, the solvent is typically bought at 99.9 atom % deuterium which means that there is a very small amount of acetone-d5 (CD3 - CO - CD2H). The - CD2H group will show up as a positive signal. Both 13C and 13C DEPT spectra are shown below for "acetone-d6". In the 13C spectrum in the bottom trace, one can see the expected 1:3:6:7:6:3:1 septet for a spin I = 1/2 nucleus coupled to three equivalent spin I = 1 nuclei. In the DEPT spectrum in the upper trace one sees only the small fraction of protonated carbons and the spectrum is a 1:2:3:2:1 quintet resulting from a spin I = 1/2 nucleus coupled to two equivalent spin I = 1 nuclei. One can also see the isotope effect of 0.254 ppm between the two isotopomers. Although the quintet is also present in the 13C spectrum, it is not seen simply because it is too small in comparison to the septet. 
Thursday, April 17, 2008
More on Long Dipolar Dephasing Delays
Further to the previous post on long dipolar dephasing delays and the application of a 180 degree 13C pulse, the figure below shows the phasing problems present with the use of long dephasing delays and how they are avoided by using a 180 degree 13C pulse at the midpoint of the dephasing delay.
Wednesday, April 16, 2008
How Much Sample Do I Need to Get a Solid State MAS NMR Spectrum?
Synthetic chemists frequently ask how much sample they need in order to get a solid state magic angle spinning spectrum. The answer is not simple and of course depends on what nucleus is to be observed, the field strength, what type of NMR experiment is needed and at what spinning speed the rotor must be set. The signal-to-noise ratio is directly proportional to the quantity of sample, so in principle, the larger the rotor, the higher the signal-to-noise ratio. This would be true if all rotors were able to spin at the rate needed for the experiment. Unfortunately, this is not the case and we are often limited by how fast we can spin the sample. The smaller the rotor - the faster the maximum available spinning speed. Often, one wants to maximize the quantity of sample (large rotors) yet employ the fastest possible spinning speeds (small rotors). A compromise must be made. The picture below (courtesy of Victor Terskikh) nicely shows the sizes of the common Bruker MAS rotors and gives an idea of the volumes of solid needed to fill the rotors.
From left to right the diameters are 7 mm, 4 mm, 3.2 mm, 2.5 mm and 1.3 mm. All of these rotor sizes are available at the National Ultrahigh Field NMR Facility for Solids. the maximum spinning speeds from left to right are 8 kHz, 18 kHz, 23 kHz, 35 kHz and 70 kHz, respectively. At the NMR Facility on the Ottawa U campus, the 7 mm, 4 mm and 2.5 mm sizes are available with maximum spinning speeds of 7 kHz, 15 kHz and 30 kHz, respectively.
Tuesday, April 15, 2008
Dipolar Dephasing with Long Dephasing Delays
The combination of cross polarization with dipolar dephasing is very useful as an assignment tool in 13C CPMAS spectra. The technique works well with dephasing delays of 40 to 60 microseconds when low MAS spinning speeds ( 2 - 5 kHz) are employed, as these speeds are insufficient to effectively average the heteronuclear dipolar coupling interaction. At higher fields, faster MAS rates are often needed to avoid an undue number of spinning sidebands. At faster spinning speeds the heteronuclear dipolar coupling interaction is more effectively averaged and much longer dephasing delays (hundreds of microseconds) must be used. The use of such long delays makes phasing the NMR spectrum impossible. In these cases a 180 degree 13C pulse can be applied at the midpoint of the dephasing delay to refocus the 13C chemical shifts and permit proper phase correction. This scheme is shown below.
Monday, April 14, 2008
Tuning and Matching an NMR Probe
The circuits in NMR probes are essentially band pass filters. The properties of the band pass filter are determined by the values of capacitance for the capacitors in the circuit and the inductance of the sample coil. One is usually able to vary the capacitance of the tuning and matching capacitors to optimize the probe for the experiment at hand. The adjustments are typically made by turning rods that extend from the bottom of the NMR probe. These rods are often labelled "T" for tune amd "M" for match. Changing the tuning capacitor will shift the band of the bandpass filter and is therefore used to tune the probe from on frequency (or nucleus) to another. This is much like tuning your radio to a particular radio station. The tuning control of a radio is also a variable capacitor. Changing the matching capacitor changes the efficiency of the band pass filter. These two capacitors are not independent and they must be adjusted iteratively to optimize the NMR probe. Proper tuning and matching are essential in getting high quality NMR results. The figure below shows the response on an oscilloscope for an NMR probe connected to a sweep generator. One can think of these plots as frequency on the horizontal axis vs efficiency (or match to 50 Ohms) on the vertical axis. The horizontal line represents a 50 Ohm reference. The first panel shows a probe that is well matched and tuned. The second panel shows a probe that is properly matched but poorly tuned and the third panel shows a probe that is properly tuned but poorly matched. 
Friday, April 11, 2008
Phasing 2D NOESY Data in TOPSPIN
In order to have a high quality 2D NOESY spectrum, both the F2 domain (rows) and F1 domain (columns) must be phased. After Fourier transforming your data with the xfb command, phase it by following these steps:
Phasing the rows manually
1. Click the interactive phase correction icon in the top symbolic menu bar.
2. Position the cross hair cursor on a diagonal peak near the top of the spectrum and then click the right mouse button. From the sub-menu that appears, click the add option.
3. Repeat step 2 for a prominent row near the center of the spectrum and for a prominent row near the bottom of the spectrum.
4. Click on the blue R icon in the data window. You should now see the three rows you have chosen on the screen with a vertical red cursor on the largest peak.
5. Click and hold down the left mouse button on the 0 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonance at the red cursor is in phase. (Remember that for the NOESY spectra of small molecules the diagonals and off-diagonals should be opposite in phase).
6. Click and hold down the left mouse button on the 1 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonances away from the red cursor are in phase.
7. Click the save and return icon in the data window. You will be returned to the NOESY spectrum and the rows will now be phased.
Phasing the columns manually
8. Click on the blue C icon in the data window. You should now see the three columns you have chosen on the screen (displayed horizontally) with a vertical red cursor on the largest peak.
9. Click and hold down the left mouse button on the 0 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonance at the red cursor is in phase. (Remember that for NOESY spectra of small molecules the diagonals and off-diagonals should be opposite in phase).
10. Click and hold down the left mouse button on the 1 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonances away from the red cursor are in phase.
11. Click the save and return icon in the data window. You will be returned to the NOESY spectrum and the columns will now be phased.
12. Click the return icon to exit the interactive phase correction routine.
Rather than phasing the rows manually, you can also phase them automatically by doing the following: (note that you will usually achieve better phase correction by phasing manually)
Phasing the rows automatically
1. Click on the ProcPars tab and under phase correction make sure that PH_mod is set to pk in both the F2 and F1 domains.
2. Click the Spectrum tab to return to the spectrum.
3. You can autophase the rows by typing calcphhomo.
4. Type xfb to Fourier trannsform you data. The rows should now be phased.
5. Phase the columns manually as described above.
The figure below shows a NOESY spectrum with both the rows and columns out of phase in the top left panel, the rows phased and the columns out of phase in the top right panel and both the rows and columns phased in the bottom panel. In this figure red is negative and black is positive.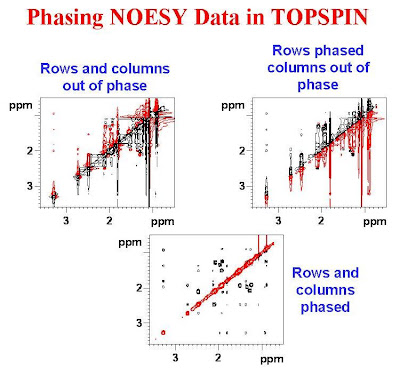
Phasing the rows manually
1. Click the interactive phase correction icon in the top symbolic menu bar.
2. Position the cross hair cursor on a diagonal peak near the top of the spectrum and then click the right mouse button. From the sub-menu that appears, click the add option.
3. Repeat step 2 for a prominent row near the center of the spectrum and for a prominent row near the bottom of the spectrum.
4. Click on the blue R icon in the data window. You should now see the three rows you have chosen on the screen with a vertical red cursor on the largest peak.
5. Click and hold down the left mouse button on the 0 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonance at the red cursor is in phase. (Remember that for the NOESY spectra of small molecules the diagonals and off-diagonals should be opposite in phase).
6. Click and hold down the left mouse button on the 1 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonances away from the red cursor are in phase.
7. Click the save and return icon in the data window. You will be returned to the NOESY spectrum and the rows will now be phased.
Phasing the columns manually
8. Click on the blue C icon in the data window. You should now see the three columns you have chosen on the screen (displayed horizontally) with a vertical red cursor on the largest peak.
9. Click and hold down the left mouse button on the 0 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonance at the red cursor is in phase. (Remember that for NOESY spectra of small molecules the diagonals and off-diagonals should be opposite in phase).
10. Click and hold down the left mouse button on the 1 icon in the data window. While holding down the left mouse button, drag the mouse up and down until the resonances away from the red cursor are in phase.
11. Click the save and return icon in the data window. You will be returned to the NOESY spectrum and the columns will now be phased.
12. Click the return icon to exit the interactive phase correction routine.
Rather than phasing the rows manually, you can also phase them automatically by doing the following: (note that you will usually achieve better phase correction by phasing manually)
Phasing the rows automatically
1. Click on the ProcPars tab and under phase correction make sure that PH_mod is set to pk in both the F2 and F1 domains.
2. Click the Spectrum tab to return to the spectrum.
3. You can autophase the rows by typing calcphhomo.
4. Type xfb to Fourier trannsform you data. The rows should now be phased.
5. Phase the columns manually as described above.
The figure below shows a NOESY spectrum with both the rows and columns out of phase in the top left panel, the rows phased and the columns out of phase in the top right panel and both the rows and columns phased in the bottom panel. In this figure red is negative and black is positive.
Wednesday, April 9, 2008
Electronics for Simple Single Frequency NMR Probes
For good reason, many NMR users find the electronics in an NMR probe daunting as they can be quite complicated, depending on the configuration of the probe (double resonance, triple resonance etc ...). The electronics in a simple single frequency NMR probe, on the other hand, are very simple and can be built on the bench in a matter of minutes. A single frequency probe can have the following tank circuit with only two capacitors and a coil. Of course, all of the components must be nonmagnetic.
CT is the tuning capacitor. It is used to tune the circuit to a specific frequency (i.e. from one nucleus to another). CM is the matching capacitor. It is used to alter the quality factor of the circuit. L is the coil in which the sample is placed. It may be a solenoid or Helmholtz coil. The inductance of the coil is an important property to consider in achieving the desired tuning range of the probe . The coil must be effective at both transmitting rf to the sample as well as detecting the small NMR signal from the sample.
CT is the tuning capacitor. It is used to tune the circuit to a specific frequency (i.e. from one nucleus to another). CM is the matching capacitor. It is used to alter the quality factor of the circuit. L is the coil in which the sample is placed. It may be a solenoid or Helmholtz coil. The inductance of the coil is an important property to consider in achieving the desired tuning range of the probe . The coil must be effective at both transmitting rf to the sample as well as detecting the small NMR signal from the sample.
Tuesday, April 8, 2008
The Field Dependence of the Second Order Quadrupolar Interaction in Solids
Most of the NMR active isotopes in the periodic table are quadrupolar, so it is understandable that there is a great deal of interest in obtaining NMR data for quadrupolar nuclei. In the solid state, one typically observes the central transition for spin I = n/2 nuclei. This transition is unaffected by the first order quadrupolar interaction however it is affected by the second order quadrupolar interaction. Magic angle spinning only partially averages the second order quadrupolar interaction and as a result one observes a broadened resonance of the central transition in MAS spectra. In favorable circumstances, the shape of this resonance can be used get information about the quadrupolar coupling and chemical shift tensors. The second order quadrupolar interaction is inversely proportional to the strength of the magnetic field so narrower lines (and therefore higher resolution) are obtained at higher field strengths. This accounts for all of the interest in ultrahigh-field NMR facilities for solids such as the one in Ottawa. The figure below shows the 27Al (I = 5/2) MAS spectrum of kaolinite obtained at three different field strengths, plotted on the same chemical shift scale. In addition to the narrowing of the resonance at higher fields, note that the "center of gravity" of the resonance also shifts with field. The "center of gravity" of the resonance approaches the true chemical shift only in the high field limit and therefore should not be reported in the literature as a "chemical shift". 
Friday, April 4, 2008
Getting ASCII Data from TOPSPIN
People often want to have an ascii file representing there NMR spectrum which can easily be imported into a spreadsheet or other program. This is easily done in Bruker's TOPSPIN program by following the steps below.
1. Display the region of the spectrum in which you are interested.
2. Right click inside the window containing the NMR spectrum.
3. Click Display Region to... from the sub-menu.
4. Highlight the checkbox, A text file for use with other programs and click the OK button.
5. Enter the desired file name and destination directory in the Please specify destination box and then click the OK button.
Your file should look something like the following:
# File created = Friday, April 4, 2008 11:33:45 AM EDT
# Data set = glenn_carborane 1 1 c:\av500data guest
# Spectral Region:
# LEFT = 4.2933867137891 ppm. RIGHT = 0.0599077215877548 ppm.
#
# SIZE = 18650 ( = number of points)
#
# In the following ordering is from the 'left' to the 'right' limits!
# Lines beginning with '#' must be considered as comment lines.
#
7250.8125
-20.265625
.
.
.etc....
The y values (intensities) are contained in the data column. This file is easily imported into any spreadsheet. The information in the header (i.e. the chemical shift limits and the total number of points) will allow the easy preparation of the x (i.e. chemical shift) data column.
1. Display the region of the spectrum in which you are interested.
2. Right click inside the window containing the NMR spectrum.
3. Click Display Region to... from the sub-menu.
4. Highlight the checkbox, A text file for use with other programs and click the OK button.
5. Enter the desired file name and destination directory in the Please specify destination box and then click the OK button.
Your file should look something like the following:
# File created = Friday, April 4, 2008 11:33:45 AM EDT
# Data set = glenn_carborane 1 1 c:\av500data guest
# Spectral Region:
# LEFT = 4.2933867137891 ppm. RIGHT = 0.0599077215877548 ppm.
#
# SIZE = 18650 ( = number of points)
#
# In the following ordering is from the 'left' to the 'right' limits!
# Lines beginning with '#' must be considered as comment lines.
#
7250.8125
-20.265625
.
.
.etc....
The y values (intensities) are contained in the data column. This file is easily imported into any spreadsheet. The information in the header (i.e. the chemical shift limits and the total number of points) will allow the easy preparation of the x (i.e. chemical shift) data column.
Thursday, April 3, 2008
1H NMR with 11B Decoupling
Boron has two isotopes with spin. 11B has a spin I = 3/2 with a natural abundance of 80.42% while 10B has a spin I = 3 with a natural abundance of 19.58%. The 1H NMR signals of protonated boron compunds are often quite complicated due to resolved or partially resolved J coupling between the protons and both 11B and 10B. A signal for a proton coupled to a single boron would be the sum of two multiplets (a 1:1:1:1 quartet from 11B and a 1:1:1:1:1:1:1 septet from 10B) in a ~ 4:1 ratio. Proton NMR spectra exhibiting boron coupling can be simplified considerably with the application of 11B decoupling. In such a spectrum, the signals for the protons coupled to 11B collapse into singlets while the signals for the protons coupled to 10B remain as multiplets. However, since 10B is only 19.58% abundant and the intensity is split into a multiplet, these signals are a tolerable contribution at the base of the narrower 1H signals. The figure below shows a standard 1H NMR spectrum and a 1H spectrum with 11B decoupling for ortho-carborane (C2B10H12). The protons attached to the carbon atoms account for the sharp signal at high frequency while the protons attached to boron account for the rest of the spectrum. One can see the dramatic simplification in the spectrum with 11B decoupling. One can also see the small contribution from the protons coupled to 10B in the baseline. The 1H [11B] spectrum confirms that there are 4 chemical shift inequivalent types of boron bearing protons which allows for an easy analysis of 1H - 11B J coupling in the standard 1H spectrum (color coded in the figure). This analysis would be difficult or impossible in the absence of the 1H [11B] data.
Thank you to Dominique Duguay for providing the sample for the figure.
Thank you to Dominique Duguay for providing the sample for the figure.
Wednesday, April 2, 2008
Finding "Lost" Deuterated 13C Signals
Several times a year I will get a question like, "I just deuterated my compound and now I can't see the 13C NMR signals for the deuterated carbons - why not?" There are at least three reasons why seeing these signals may be difficult. First of all, the principal relaxation mechanism for protonated carbons is the dipolar interaction between the 13C and the attached protons. When the protons are replaced by deuterium the dipolar mechanism for relaxation is much less efficient and the T1 for the deuterated carbon may be more than an order of magnitude greater than that of the comparable protonated carbon. The long T1 for the deuterated carbon may mean that it will be saturated when using the "standard parameters" and either not show up or be of much reduced intensity. Secondly, the intensity of the carbon signal will be split into a multiplet due to 13C - 2H J coupling depending on how many deuterons are attached to the carbon (1:1:1 triplet for CD, 1:2:3:2:1 pentet for CD2 and 1:3:6:7:6:3:1 septet for CD3). This means that the intensity of the proton decoupled CHn singlet will be spread over all of the lines of the 13C - 2H J multiplet with a corresponding large decrease in signal-to-noise ratio for any one of the lines of the multiplet compared to the proton decoupled singlet for the CHn carbon. Thirdly, there is almost no nuclear Overhauser enhacement for deuterated carbons as there are no directly bound protons. This means that if proton decoupling is applied both during the acquisition and the recycle delay, the protonated carbon signals will grow at a much faster rate than the deuterated carbon signals as the number of scans is increased. In the figure below is an example. The lower panel shows the partial spectrum of a protonated organic molecule using proton decoupling during both the acquisition time and the 2 second recycle delay. The middle panel shows the similar spectrum of a partially deuterated molecule. Note that the deuterated carbon signals are "lost". The top panel shows a spectrum acquired with the same number of scans as that in the middle panel but with a 60 second recycle delay and decoupling only during the acquisition time (i.e. not during the recycle delay). Acquirng the data in this way eliminates the problems of long relaxation times and differential nOe's. In this spectrum one can see the deuterated carbon signals however the signal-to-noise ratio is lower than those for the protonated carbons due to the splitting from 13C - 2H J coupling. In this particular case also, a further loss in signal-to-noise ratio results because the nitrogen bearing carbon is also broadened due to partial self decoupling of the 14N from the 13C.
Bottom line - collect your data over a long period of time with a long recycle delay and inverse gated proton decoupling.
Thank you to David Lapointe for providing the samples for the figure.

Bottom line - collect your data over a long period of time with a long recycle delay and inverse gated proton decoupling.
Thank you to David Lapointe for providing the samples for the figure.
Tuesday, April 1, 2008
New Solids NMR Equipment for Ottawa U.
The University of Ottawa will take delivery of a new Bruker AVANCE III 400 NMR spectrometer for solids this summer. This instrument will be installed in MacDonald Hall, room 012 with the Bruker AVANCE 500. In addition to the new 400, the 14 year old console for the ASX 200 spectrometer for solids will be replaced with a new Bruker AVANCE III 200 console. Also this summer, the Bruker AVANCE 500 spectrometer will be upgraded from XWINNMR 3.5 to TOPSPIN 2.0. Congratulations to Dr. David Bryce for his successful CFI proposal.
Subscribe to:
Posts (Atom)
